Help:
Data Input Explanation (Step 1)
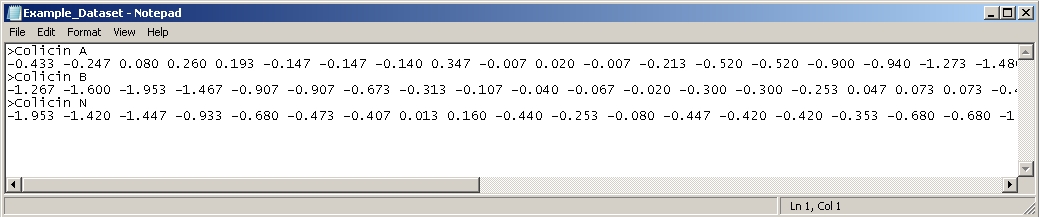
Multi-profile comparison: Multiple data profiles should be entered as either a set of single space-delimited floating-point numbers, or as a set of non-delimited amino acid sequences. In either case, the sequences are separated by a return and a ">" sign, and an optional protein name. Every dataset will be compared against every other dataset and the graphical and numerical results will be returned. The datasets should look like FASTA format, the only difference being the added option of space delimited floating-point numbers in addition to non-delimited amino acids. See below for an example.
Example:
Parameter Explanation (Step 2)
Probability Model:
HePCaT will return a statistical significance (a p-value) with your alignments based on the specific type of data submitted. The probability models are pre-computed, therefore, if your dataset is not one of the above options, a statistical significance cannot be returned. In this case, choose "None".
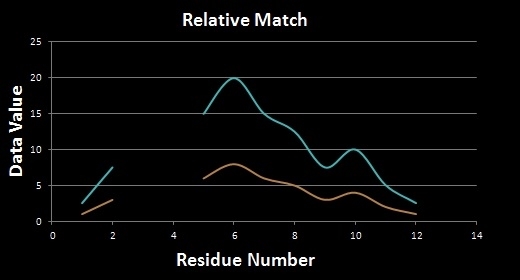
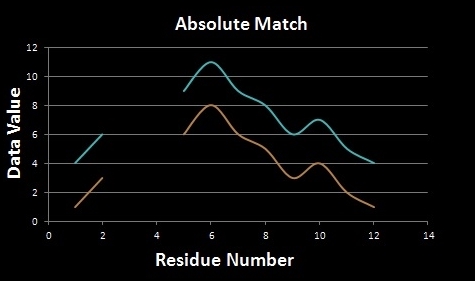
Local Scaling:
Local Scaling allows for HePCaT to match relative shapes, as opposed to absolute shapes. Relative shape matching is useful for finding patterns that reflect a similar behavior with proportional amplitudes. Absolute shape matching is useful in finding the same shape in both behavior and magnitude, such as a repeated template in the dataset. See the illustration below:
Window Size:
This parameter determines the unit length of fragments that contribute to the global alignment. The total alignment is composed of fragments of data that match well to each other. If the window size is 5, these fragments will be composed of 5 data points each.
Cutoff:
This value determines the stringency of your matches. The higher the cutoff, the more similar your matches must be to be considered in the final alignment. For example, a cutoff of 0.80 means that the fragment matches will only be considered if they are 80% identical.
Max. Gap Length:
Gaps allow for breaks in global alignments, and this parameter determines the maximum length of those gaps. Gaps are useful as they allow for flexibility in fragment matching.